


济南顺奇净化工程有限公司
电话(传真):0531-68824415
手 机:13854165330
Q Q:340095748
联系人:张经理
邮 编:250024
邮 箱:340095748@qq.com
地 址:山东省济南市天桥区新徐居委会黄河建邦大桥西侧1-6号
原料药一般由化学合成、DNA重组技术、发酵、酶反应或从天然物质提取制得,它是加工药物制剂的主要原料, 原料药分无菌原料药和非无菌原料药。前者是要按规定进行法定的无菌检查,后者则不需要作此检查。
GMP(2010)对非无菌原料药的精、烘、包操作工序的暴露环境推荐采用无菌药品的D级区标准。
所谓“精”,即精制,包括精滤、结晶、分离、检验等工序。
所谓“烘“,即干燥,包括干燥、粉碎、混粉及检验等工序。
所谓“包”,即包装,包括包装材料的处理和包装等工序。
原料药精、烘、包各工序的特点是:
(1)精滤对法定要无菌检查的,对无菌药品确定环境级别;不需无菌检查的,则只要求D级环境。
(2)结晶要求与精滤相同。
(3)分离注意分离有机溶媲或有气妹的品种时,应采取措施,防止溶媒或气味在室内扩散。
(4)干燥(俗称烘干)尽可能采用干燥、混粉一次完成的设备;干燥时所用的空气应经净化处理,达到与生产环境同等洁净程度;尾气需经捕集、除尘后再排放。
(5)粉碎和过筛应有局部防尘或吸尘装置。
(6)检验。
(7)包装材料处理注意直接接触药品的包装材料应按适当方法清洁消毒,其中无菌原料药所用的内包材要用经过过滤的注射用水冲洗,并在4h内灭菌,24h内使用,且均应专柜贮存在与包装同等洁净度级别的贮存室内。
(8)包装有换批生产时,注意做好清场工作。
图9-1是非无菌原料药工艺示意图。
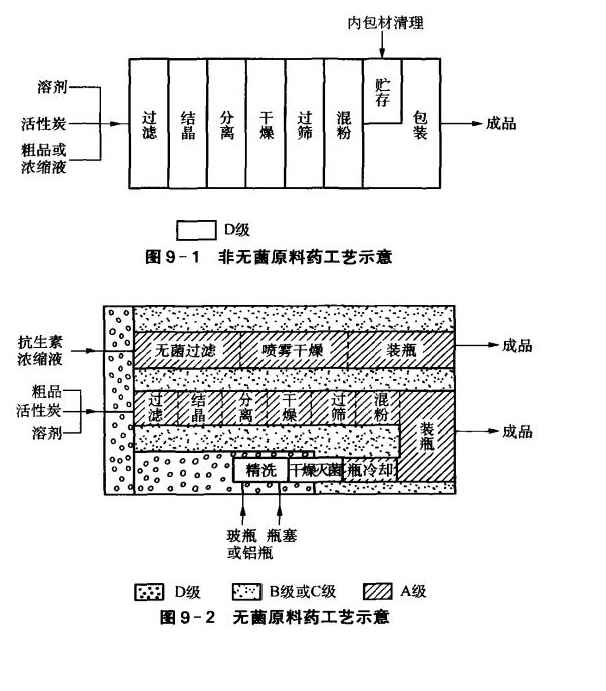
图9-2是无菌原料药工艺示意图。
(9)厂房设计GMP规定原料药的质量标准中有热原或细菌内毒素等检验项目的,厂房的设计应特别注意防止微生物污染,如应根据工艺要求设定相应厂房的洁净度级别。
质检实验室通常应与生产区分开。当生产操作和检验结果之间无互相不利影响时,中间控制实验室可设在生产区内。
GMP(2010)的术语定义指出中间控制“也称过程控制,指为确保产品符合有关标准,生产中对工艺过程加以监控,以便在必要时进行调节而做的各项检查。”当然,中间控制操作不得给药品带来质量风险。
(10)设备可用同一设备生产,但应有防止交叉污染的措施,应有适当间隔。难以清洁的设备或部件应专用。在无菌操作条件下添加细胞基质、培养基、缓冲液和气体,应当采用密闭或封闭系统。
(11)清洗更换批次或品种生产前,必须对设备彻底清洗。
(12)发酵工艺采用发酵工艺生产原料药应根据生产步骤和生产条件(敞口、密闭或封闭系统)确定环境控制标准。初始容器接种、转种或加料(培养基、缓冲液)使用敞口容器操作时,如微生物污染可能危及原料药质量时,敞口容器应置于适当的控制环境中。 需在无菌操作条件下添加细胞基质、培养基、缓冲液和气体时,应采用密闭或封闭系统。
(13)容器对可重复使用的容器应行清洁并去除或涂毁其原有标签。
