


жµОеНЧй°Їе•ЗеЗАеМЦеЈ•з®ЛжЬЙйЩРеЕђеПЄ
зФµиѓЭпЉИдЉ†зЬЯпЉЙпЉЪ0531-68824415
жЙЛ жЬЇпЉЪ13854165330
Q QпЉЪ340095748
иБФз≥їдЇЇ:еЉ†зїПзРЖ
йВЃ зЉЦ:250024
йВЃ зЃ±:340095748@qq.com
еЬ∞ еЭА:е±±дЄЬзЬБжµОеНЧеЄВ姩氕еМЇжЦ∞еЊРе±ЕеІФдЉЪйїДж≤≥еїЇйВ¶е§Іж°•и•њдЊІ1-6еПЈ
еЬ®
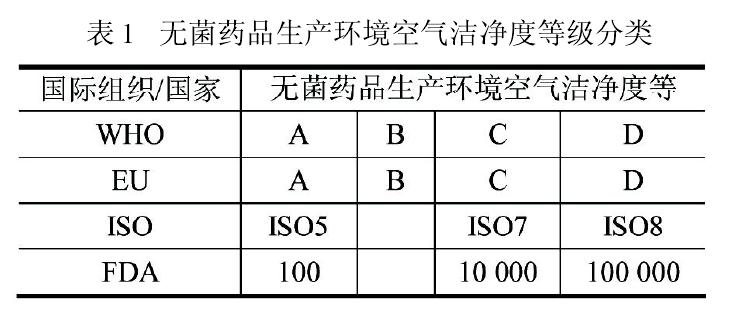
еЕ≥дЇОињЩдЄ§зІНзЬЛдЉЉдЄНеРМзЪДж®°еЉПпЉМжђІзЫЯ GMP жМЗеНЧпЉИ2009пЉЙеЉХи®АдЄ≠жЬЙдЄАжЃµиАРдЇЇеѓїеС≥зЪДи°®ињ∞пЉЪвАЬжЬђжМЗеНЧжЧ†жДПжИРдЄЇдїїдљХжЦ∞ж¶ВењµжИЦжЦ∞жКАжЬѓеПСе±ХзЪДйЪЬзҐНпЉМе¶ВжЮЬйАЪињЗй™МиѓБеєґиѓБжШОжЙАзФ®жЦєж≥ХиГљиЊЊеИ∞иЗ≥е∞СдЄОжЬђжМЗеНЧжЙАињ∞жЦєж≥Хз≠ЙдїЈзЪДиі®йЗПдњЭиѓБж∞іеє≥пЉМдєЯеЇФдЇИдї•иЃ§еРМпЉИTheGuide is not intended to place any restraint upon thedevelopment of any new concepts or new technologieswhich have been validated and which provide a level of Quality Assurance at least equivalent to those set out in this GuideпЉЙвАЭгАВеРМжЧґпЉМеЃГињШжШОз°Ѓи°®иЊЊвАЬиНѓеУБе§ЪеєіжЭ•жМЙ GMP и¶Бж±ВзФЯдЇІпЉМж≤°жЬЙжЙІи°М ISO зЪДж†ЗеЗЖгАВдЉБдЄЪеПѓиЗ™и°МеЖ≥еЃЪйЗЗзФ®ISOж†ЗеЗЖдљЬдЄЇеЃЮжЦљеИґиНѓйҐЖеЯЯиі®йЗПдљУз≥їзЪДдЄАзІНжЙЛжЃµпЉИThe manufacture of medicinal products has for many years taken place in accordance withguidelines for GMP and the manufacture of medicinalproducts is not implement by ISO standards. Harmo-nized standards as adopted by the ISO may be used atindustryвАЩs discretion as a tool for implementing a quality system in the pharmaceutical sectorпЉЙвАЭгАВ
еРМж†ЈпЉМFDA зЪДжМЗеНЧжЦЗдїґдєЯжЬЙз±їдЉЉеЖЕеЃєзЪДи°®ињ∞пЉМиґ≥иІБињЩдЄ§зІНж®°еЉПеЬ®еЫљйЩЕеИґиНѓи°МдЄЪзЪДдЇТиЃ§еТМеЕ±еЃєгАВжђІзЫЯGMPжМЗеНЧдЄОISO14644.4 еЬ®жОІеИґжЦєж≥ХдЄКзЪДеЈЃеЉВпЉМеєґдЄНиѓіжШОеЃГдїђеЬ®жОІеИґж∞іеє≥дЄКзЪДеЈЃиЈЭгАВеЫљйЩЕеИґиНѓдЉБдЄЪеЬ®еЃЮжЦљжЧґеПѓиЗ™и°МйАЙзФ®пЉМжЧ†й°їеОЪж≠§иЦДељЉпЉМеП™и¶БиЃ§зЬЯжЙІи°МйГљиГљиЊЊеИ∞еРМж†ЈзЪДиі®йЗПдњЭиѓБж∞іеє≥гАВеЫ†ж≠§пЉМзЊОеЫљеИґиНѓдЉБдЄЪдЇІеУБжЧ†йЬАжМЙжђІзЫЯзЪДABCDж®°еЉПжЦєеПѓињЫеЕ•жђІзЫЯеЫљеЃґпЉЫеРМж†ЈжђІзЫЯеИґиНѓдЉБдЄЪдЇІеУБдєЯж≤°жЬЙењЕи¶БжФєжИРFDA зЪДACD ж®°еЉПжЙНиГљињЫеЕ•зЊОеЫљеЄВеЬЇгАВеПѓжШѓпЉМељУињЩдЄ§й°єеЫљйЩЕж†ЗеЗЖжЉФеПШжИРжИСеЫљзЪДж≥ХиІДеТМж†ЗеЗЖжЧґпЉМжИСеЫљзЪДеИґиНѓдЉБдЄЪе∞±ж≤°жЬЙињЩдєИеєЄињРгАВеЫ†дЄЇжИСеЫљжЦ∞зЙИGMPеЬ®з≠ЙжХИйЗЗзФ®жђІзЫЯGMPжМЗеНЧжЧґпЉМеИ†йЩ§дЇЖжђІзЫЯGMPжМЗеНЧзЪДињЩжЃµвАЬжПРз§ЇвАЭгАВжЦ∞зЙИ GMP еПСеЄГеЙНпЉМжИСеЫљдї•еЊА GMPдЄ≠жЧ†иПМиНѓеУБзФЯдЇІзОѓеҐГзЪДз©Їж∞ФжіБеЗАеЇ¶ж†ЗеЗЖеЯЇжЬђйЗЗзФ®ACD ж®°еЉПгАВеЃҐиІВдЄКпЉМжИСеЫљеОЖзЙИ GMP еЖЕеЃєеТМдЉБдЄЪжЙІи°МеКЫеЇ¶йГљдЄОеЫљйЩЕи¶Бж±Ве≠ШеЬ®дЄАеЃЪеЈЃиЈЭпЉМињљеЕґеОЯеЫ†йФЩзїЉе§НжЭВпЉМжИСеЫљжЧ†иЃЇеЬ®иЃ§зЯ•зРЖењµгАБзїЉеРИеЫљеКЫгАБжКАжЬѓж∞іеє≥з≠ЙжЦєйЭҐйГљжЧ†ж≥ХдЄОеЫље§ЦеПСиЊЊеЫљеЃґзЫЄжѓФпЉМеН≥дљњзЕІжРђзЕІе•ЧеЫље§Ц GMPпЉМдєЯдЄНиГљиµЈеИ∞зЂЛзЂњиІБељ±зЪДжХИжЮЬгАВе¶ВжЮЬжЦ∞зЙИ GMP иГљж†єжНЃеЫљжГЕеРЄзЇ≥жђІзЫЯ GMP жМЗеНЧзЪДеЃљеЃєпЉМе§Іе§ЪжХ∞дЉБдЄЪеПѓеЬ®еОЯжЬЙеЯЇз°АдЄКињЫи°Ме°Ђеє≥и°•йљРзЪДжФєйА†пЉМиАМжЧ†йЬАе¶ВдїКдЉ§з≠ЛеК®й™®еЉПзЪДйЗНеїЇгАВињЩжЧ†иЃЇеѓєеЫљеЃґињШжШѓеѓєдЉБдЄЪпЉМйГљжШѓеАЉеЊЧжЈ±жАЭзЪДе§ІдЇЛгАВ
жЦ∞зЙИ GMP жШѓеЉЇеИґжЙІи°МзЪДеЫљеЃґи°МжФњж≥ХиІДпЉМжШѓеИґиНѓдЉБдЄЪиЃ§иѓБж£АжЯ•зЪДеФѓдЄАж†ЗеЗЖпЉМеЊЧдЄНеИ∞иЃ§иѓБиѓБдє¶пЉМдЉБдЄЪењЕй°їеЕ≥еБЬеєґиљђпЉЫиАМB/T25915 жШѓеЫљеЃґжО®иНРж†ЗеЗЖпЉМеПИжЧ†зЫЄеЇФж£АжЯ•йҐБиѓБжЙЛжЃµпЉМиЗ™зДґжЧ†ж≥ХдЄОжЦ∞зЙИGMPзЫЄжПРеєґиЃЇгАВеЗЇзО∞е¶Вж≠§йЧЃйҐШпЉМеєґдЄНиѓіжШОињЩдЄ§й°єж†ЗеЗЖзЪДж∞іеє≥еЈЃиЈЭпЉМиАМжШѓеЫљеЃґи°МжФњеИґеЇ¶гАБжЭГеКЫдЄКзЪД姱谰пЉМйЬАи¶БзФ±жФњеЇЬжЬЙеЕ≥йГ®йЧ®йЗЗеПЦжО™жЦљдЇИдї•еЉ•еРИдњЃи°•гАВGB/T 25915.4пЉИ2010пЉЙпЉНиЃЊиЃ°гАБеїЇйА†гАБеРѓеК®пЉМеН≥ ISO14644.4 зЪДйЩДељХ BпЉМжПРеЗЇдЇЖжЧ†иПМдЇІеУБеЬ®йҐЧз≤ТзЙ©еТМеЊЃзФЯзЙ©еПЧжОІзЪДжіБеЗАеМЇеЖЕжЧ†иПМзБМи£ЕзЇњдЄКзБМи£ЕзЪДз©Їж∞ФжіБеЗАеЇ¶з≠ЙзЇІи¶Бж±ВпЉИи°® 2 пЉЙгАВ
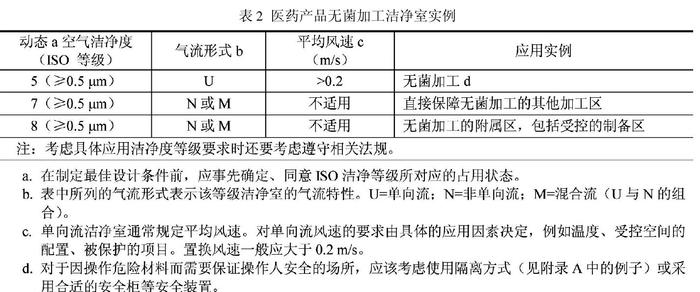
и°®2дЄ≠еАЉеЊЧжИСдїђж≥®жДПзЪД пЉМдЄАжШѓз©Їж∞ФжіБеЗАеЇ¶з≠ЙзЇІзЪДеН†жЬЙзКґжАБеЇФеЬ®иЃЊиЃ°еЙНзФ±дЄЪдЄїеТМиЃЊиЃ°дЇЇеСШдЇЛеЕИеХЖиЃЃз°ЃеЃЪгАВињЩжШѓ ISO14644 з≥їеИЧж†ЗеЗЖеЬ®еЃЪдєЙвАЬеК®жАБвАЭгАБвАЬйЭЩжАБвАЭжЧґзЪДдЄАиіѓдЄїеЉ†пЉМеЫ†дЄЇеРДзІНеН†зФ®зКґжАБйГљжЬЙеЃГзЪДдљњзФ®дїЈеАЉпЉМдЄНиГљзЃАеНХеЬ∞дї•дљњзФ®дљХзІНвАЬеН†зФ®зКґжАБвАЭжЭ•иѓДеИ§ж†ЗеЗЖзЪДе≠∞йЂШе≠∞дљОгАВеПЧжЧ†иПМиНѓеУБзБМи£ЕзЇњдЄКдЇЇеСШжУНдљЬгАБиЃЊе§ЗжАІиГљгАБзФЯдЇІињРи°Мз≠ЙеЫ†зі†ељ±еУНпЉМеНХеРСжµБзљ©дЄЛзЪДз©Їж∞ФжіБеЗАеЇ¶еЊАеЊАйЪЊдї•жµЛеЗЖпЉМдЄЇз°ЃдњЭжµЛиѓХзКґжАБзЪДз®≥еЃЪжАІеТМеИ§жЦ≠зїУжЮЬзЪДеПѓжѓФжАІпЉМжЧ•жЬђжККжµЛиѓХи¶Бж±ВеОЯеИЩдЄКеЃЪдЄЇвАЬйЭЩжАБвАЭпЉМдєЯжЬЙеЫљеЃґиІДеЃЪеК®жАБж£АжµЛиМГеЫідЄНеМЕжЛђжУНдљЬзВєжИЦеПСе∞ШзВє
GB/T25915.4пЉИ2010пЉЙжЬЙзЭАдЄОжЦ∞зЙИ GMP еРМж†ЈзЪДжОІеИґзРЖењµпЉМйГљдЄЇдЇЖжЬЙжХИжОІеИґиНѓеУБзФЯдЇІзОѓеҐГзЪДж±°жЯУгАВеЫЊ 1 жШѓ GB/T25915.2010пЉЙзЪДе±ВеЉПж±°жЯУжОІеИґж¶ВењµгАВеЈ•иЙЇж†ЄењГеМЇпЉИдЄОзОѓеҐГеПСзФЯдЇ§дЇТдљЬзФ®зЪДеЈ•иЙЇдљНзљЃпЉЙжЙАеЬ®зЪДеМЇеЯЯдЄЇжіБеЗАеМЇпЉМжШѓжіБеЗАеЃ§дЄ≠еПЧжОІжЬАдЄ•зЪДйГ®еИЖпЉМз©Їж∞ФжіБеЗАеЇ¶з≠ЙзЇІдЄЇеНХеРСжµБ100 зЇІпЉИеН≥ISO 5 зЇІпЉМжђІзЫЯ A зЇІпЉЙгАВеЗЇдЇОзїПжµОгАБжКАжЬѓеТМињРи°Мз≠ЙжЦєйЭҐзЪДеОЯеЫ†пЉМи¶Бе∞љйЗПеЗПе∞ПжЬАйЂШжіБеЗАеЇ¶еМЇеЯЯзЪДиМГеЫігАВжіБеЗАеМЇйАЪеЄЄйЗЗзФ®еѓЖйЧ≠еЉПпЉИе¶В RABSпЉЙпЉМжИЦзФ±жіБеЗАеЇ¶иЊГдљОзЪДе§ЦйГ®еМЇеЯЯпЉНжіБеЗАеЃ§пЉИ1 дЄЗзЇІпЉМеН≥ ISO7 зЇІпЉЙеМЕеЫігАВзЫЄйВїеМЇеЯЯйЧізЪДдЇЇжµБгАБзЙ©жµБињЫеЕ•еЈ•иЙЇж†ЄењГеМЇжЧґпЉМдЉЪеҐЮеК†дЉ†жТ≠ж±°жЯУзЪДй£ОйЩ©пЉМеЫ†ж≠§еЇФзЙєеИЂж≥®жДПдЇЇжµБеТМзЙ©жµБзЪДеЄГе±АзїЖиКВдЄОзЃ°зРЖпЉМињЩе∞±жШѓе±ВеЉПж±°жЯУжОІеИґзЪДеЯЇжЬђж¶ВењµгАВ
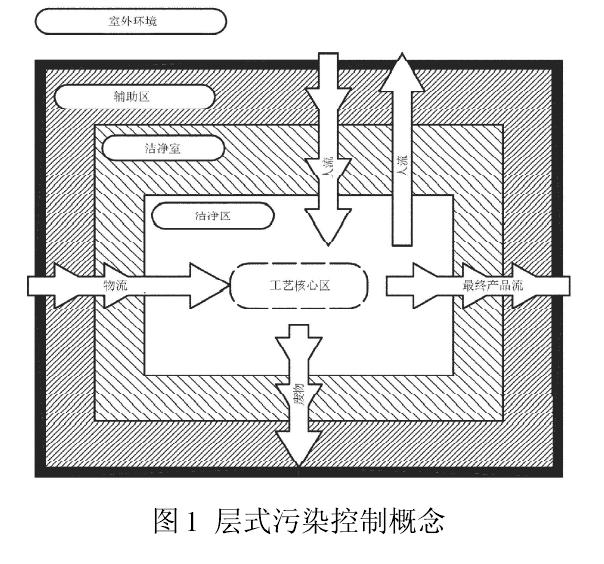
дЄО GB/T25915.4пЉИ2010пЉЙдЄНеРМзЪДжШѓжЦ∞зЙИGMPе∞ЖеМЕеЫіжіБеЗАеМЇзЪДе§ЦйГ®еМЇеЯЯпЉИжіБеЗАеЃ§пЉЙзЪДз©Їж∞ФжіБеЗАеЇ¶з≠ЙзЇІиЃЊеЃЪдЄЇBзЇІпЉМжЙАи∞УBзЇІжШѓйЭЩжАБжОІеИґжЧґзЫЄеРМдЇОA зЇІпЉИеНХеРСжµБ 100 зЇІпЉЙзЪДйЭЩжАБпЉМеК®жАБжОІеИґжЧґзЫЄеРМдЇО C зЇІпЉИ1 дЄЗзЇІпЉЙзЪДйЭЩжАБгАВGB/T 25915.4пЉИ2010пЉЙеТМжЦ∞зЙИ GMP йГљиЃ§дЄЇжЧ†иПМзБМи£ЕзЇње§ЦйГ®еМЇеЯЯпЉИжіБеЗАеЃ§пЉЙз©Їж∞ФжіБеЗАеЇ¶з≠ЙзЇІеЇФдљОдЇОеЈ•иЙЇж†ЄењГеМЇпЉИжіБеЗАеМЇпЉЙпЉМиЗ≥дЇОдљОе§Ъе∞СпЉМжШѓ1 дЄЗзЇІињШжШѓB зЇІпЉЯињЩжШѓеПѓжОҐиЃ®зЪДе≠¶жЬѓйЧЃйҐШпЉМдЄНеЇФжИРдЄЇеИ§еЃЪж†ЗеЗЖйЂШдљОзЪДеИЖзХМзЇњгАВеЫљеЖЕе§ЦеЃЮиЈµиѓБжШОпЉМиГМжЩѓиЃЊзљЃдЄЇ1 дЄЗзЇІжИЦB зЇІпЉМйГљиГљдњЭжК§жЧ†иПМиНѓеУБзЪДзФЯдЇІзОѓеҐГпЉМжИСдїђеЇФдїОеИЖжЮРељ±еУНжЧ†иПМзБМи£ЕзЇњзЪДзОѓеҐГеЫ†зі†зЭАжЙЛпЉМеѓїжЙЊжОІеИґж±°жЯУжЇРзЪДйАФеЊДпЉМжЙНиГљжЬАзїИз°ЃдњЭжЧ†иПМзФЯдЇІзОѓеҐГгАВ
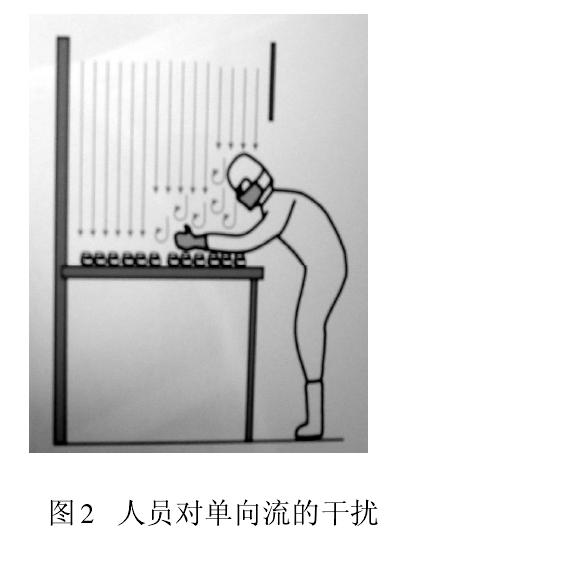
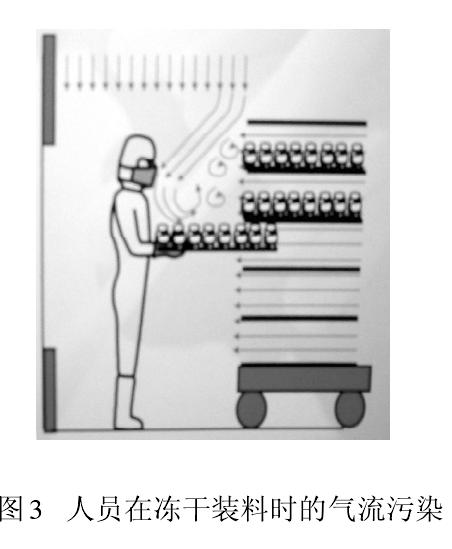
дїОдЇЇдїђеЉАеІЛжО•еПЧеєґеЇФзФ®жіБеЗАжКАжЬѓжЧґе∞±еПСзО∞пЉМдЇЇжШѓжіБеЗАеЃ§жЬАе§ІзЪДж±°жЯУжЇРгАВдї•еЊЃз≤ТеТМеЊЃзФЯзЙ©дЄЇж±°жЯУзЙ©жОІеИґеѓєи±°зЪДеИґиНѓзФЯдЇІпЉМе§ІйЗПжµЛиѓХжХ∞жНЃеТМзФЯдЇІеЃЮиЈµеСКиѓЂжИСдїђпЉМдЇЇеСШпЉИзФЯдЇІгАБж£Ай™МгАБзЃ°зРЖгАБзїідњЃпЉЙеЫ†зі†жЧ†жЧґжЧ†еИїдЄНеЬ®еє≤жЙ∞иНѓеУБзФЯдЇІгАВеЫЊ2 жШЊз§ЇдЇЇеСШеЬ®еНХеРСжµБдЄЛжУНдљЬеѓЉиЗіж∞ФжµБз≤Те≠Рж±°жЯУгАВж≠§еИїпЉМдњЭжК§жАІзЪДеНХеРСжµБеПШжИРж±°жЯУзЙ©зЪДиљљдљУпЉМзЫіжО•ељ±еУН襀дњЭжК§зЪДдЇІеУБгАВж≠§жЧґж∞ФжµБйАЯеЇ¶иґКе§ІпЉМеИЩж±°жЯУз®ЛеЇ¶иґКе§ІгАВеЫЊ3 дЄЇеЖїеє≤еЙВи£ЕжЦЩжЧґпЉМеНХеРСжµБзљ©дЄЛеЮВзЫіеТМж∞іеє≥ж∞ФжµБзЫЄдЇТеЖ≤з™БпЉМ
嚥жИРзіКжµБпЉМзіКжµБж∞ФжµБдЉЪеЬ®иЊГйХњжЧґйЧійЗМдЇІзФЯжВђжµЃж±°жЯУгАВ
дЄНи®АиАМеЦїпЉМдЇЇжШѓжЧ†иПМзБМи£ЕињЗз®ЛдЄ≠ељ±еУНжЬАе§ІзЪДзОѓеҐГеЫ†зі†гАВдЄЇж≠§пЉМеЃЮзО∞жЧ†иПМзБМи£ЕзОѓеҐГжОІеИґзЪДжКАжЬѓи¶БзВєжШѓпЉЪпЉИ1пЉЙе∞ЖдЇЇеСШдїОжЧ†иПМзОѓеҐГйЪФз¶їпЉМдїОж†єжЬђдЄКжОТйЩ§дЇЇзЪДеє≤жЙ∞пЉЫпЉИ2пЉЙйЗЗеПЦе±АйГ®йЪФз¶їжЧ†иПМзОѓеҐГ пЉИе¶ВR A B S з≠ЙпЉЙпЉЫпЉИ3 пЉЙеН≥дЊње¶Вж≠§дїНйЬАйЩРеИґдЇЇеСШеѓєжЧ†иПМдЇІеУБзЪДеє≤йҐДпЉЫпЉИ4пЉЙзФЯдЇІињРи°МдЄ≠дЄНиЃ©дЇЇеСШињЫеЕ•жЧ†иПМзОѓеҐГгАВдЄНиІ£еЖ≥дЇЇеѓєжЧ†иПМзОѓеҐГеє≤жЙ∞йЧЃйҐШпЉМе§ІеєЕеЇ¶жПРйЂШжЧ†иПМиНѓеУБзФЯдЇІзЇње§ЦйГ®еМЇеЯЯз©Їж∞ФжіБеЗАеЇ¶з≠ЙзЇІпЉМжХИжЮЬдєЯдЄНдЉЪжШОжШЊгАВ
ењЕй°їеЉЇи∞ГпЉМеМїиНѓеЈ•з®ЛиЃЊиЃ°дЄ≠з©Їж∞ФеЗАеМЦжО™жЦљеП™жШѓжОІеИґжіБеЗАеЃ§еЊЃзФЯзЙ©зЪДжЙЛжЃµдєЛдЄАпЉМињЩдЇЫжО™жЦљеП™иГљжОІеИґйАЪињЗз©Їж∞ФйАФеЊДдЉ†жТ≠зЪДеЊЃзФЯзЙ©ж±°жЯУпЉМеѓєи°®йЭҐеѓєи±°еЊЃзФЯзЙ©зЪДзФЯзЙ©ж±°жЯУдї•еПКеМЦе≠¶ж±°жЯУйГљжЧ†иГљдЄЇеКЫпЉМиНѓеУБзФЯдЇІдљњзФ®зЪДиЃЊе§ЗгАБиЃЊжЦљгАБеЃєеЩ®еЕЈгАБзЙ©жЦЩгАБеМЕи£ЕжЭРжЦЩз≠Йж≤ЊйЩДзЪДеЊЃзФЯзЙ©гАБеМЦе≠¶еУБпЉМдї•еПКзФЯдЇІгАБзЃ°зРЖдЇЇеСШжРЇеЄ¶зЪДеЊЃзФЯзЙ©пЉМеП™жЬЙйЗЗзФ®жЄЕеЬЇгАБжЄЕжіЧгАБжґИжѓТгАБзБ≠иПМз≠ЙжО™жЦљпЉМдїїдљХзЇІеИЂзЪДеЗАеМЦз©Їи∞ГйГљжЧ†ж≥ХжЫњдї£гАВ
ињСеєіжЭ•еЫље§Цеѓєз©Їж∞ФеЗАеМЦзЪДжї§иПМжХИжЮЬжПРеЗЇиі®зЦСгАВжХ∞жНЃи°®жШО 1 еП∞
еЗАжКАжЬѓиАБиЈѓпЉМе∞ЖдЉЪе§Іе§Іжµ™иієжіБеЗАеЃ§еїЇиЃЊиієзФ®еТМињРи°МжИР жЬђ гАВ
еЫљж†З GB/T 25915.4пЉИ2010пЉЙзЪДеЖЕеЃєеМЕжЛђиІДеИТеТМиЃЊиЃ°гАБеїЇйА†еТМеРѓеК®гАБж£АжµЛеТМй™МжФґгАБжЦЗдїґз≠ЙжЦєйЭҐи¶Бж±ВпЉМжґµзЫЦеЈ•з®ЛеїЇиЃЊзЪДеЕ®ињЗз®ЛпЉМеѓєжИСеЫљеМїиНѓи°МдЄЪе∞§дЄЇйЗНи¶БгАВиНѓеУБзФЯдЇІеЃЮжЦљ GMP жШѓдЄАй°єз≥їзїЯеЈ•з®ЛпЉМдЄЇиНѓеУБзФЯдЇІжПРдЊЫзђ¶еРИ GMP и¶Бж±ВзЪДеОВжИњиЃЊжЦљжШѓдЉБдЄЪеЃЮжЦљG M P зЪДеЕИеЖ≥жЭ°дїґгАВеЫ†ж≠§пЉМеМїиНѓеЈ•з®Лй°єзЫЃењЕй°їжМЙGMP и¶Бж±ВеЃЮжЦљиІДиМГзЃ°зРЖпЉМжЙНиГљдЄЇдЉБдЄЪеЃЮжЦљ GMPжПРдЊЫеПѓйЭ†дњЭйЪЬгАВињСеєіжЭ•еЫље§ЦйТИеѓєеМїиНѓеЈ•з®Лй°єзЫЃзЪДиІДиМГеМЦзЃ°зРЖпЉМдЄАй°єжЦ∞зЪДзЃ°зРЖиІДиМГвАХ вАХ GEPеЈ≤еЬ®дЄАдЇЫеПСиЊЊ
еЫљеЃґжВДзДґйЧЃдЄЦгАВGEPпЉИеМїиНѓеЈ•з®Лиі®йЗПзЃ°зРЖиІДиМГпЉЙжШѓGood Engineering Practice зЪДзЉ©еЖЩпЉМжШѓдЄОGMPйЕНе•ЧзЪДиНѓеУБзФЯдЇІзЃ°зРЖжЦЗдїґгАВеЃГзЪДеЃЧжЧ®жШѓвАЬе∞ЖеЈ≤з°ЃзЂЛзЪДеЈ•з®ЛиЃЊиЃ°жЦєж°ИеТМж†ЗеЗЖеЇФзФ®дЇОй°єзЫЃзЪДжХідЄ™зФЯеСљеС®жЬЯдЄ≠пЉМжПРдЊЫеРИйАВжИРжЬђзЪДиІ£еЖ≥жЦєж°ИвАЭгАВжИСеЫљиЗ≥дїКе∞ЪжЧ†ињЩжЦєйЭҐзЪДиІДиМГпЉМеЈ•з®Лй°єзЫЃе§Іе§ЪзФ±дЉБдЄЪеЗ≠зїПй™МиЗ™и°МзЃ°зРЖпЉМдЄїи¶БдЊЭжНЃжШѓеРДз±їзЫЄеЕ≥зЪДеЫљеЃґж†ЗеЗЖпЉМзЉЇдєПз≥їзїЯжАІгАБеНПи∞ГжАІгАВ
еЫљж†З GB/T 25915.4пЉИ2010пЉЙзЪДеПСеЄГпЉМдЄЇеЈ•з®Лй°єзЫЃиІДеИТгАБиЃЊиЃ°гАБжЦљеЈ•гАБиѓХиљ¶гАБж£АжµЛгАБй™МжФґз≠ЙзОѓиКВзЪДиІДиМГеМЦзЃ°зРЖе°Ђи°•дЇЖз©ЇзЩљпЉМеЫљж†ЗжЙАйЩДзЪДе§ІйЗПиµДжЦЩжАІйЩДељХпЉМе¶ВвАЬиЃЊжЦљзЪДй™МжФґвАЭгАБвАЬиЃЊжЦљзЪДеЄГе±АвАЭгАБвАЬеїЇйА†еТМжЭРжЦЩвАЭгАБвАЬжіБеЗАеЃ§зЪДзОѓеҐГжОІеИґвАЭгАБвАЬеЊЕйЬАжЦє/ зФ®жИЈдЄОдЊЫжЦє/ иЃЊиЃ°жЦєеХЖеЃЪзЪДи°•еЕЕжКАжЬѓи¶Бж±ВвАЭз≠ЙпЉМдЄЇй°єзЫЃзЪДеЈ•з®ЛиЃЊиЃ°гАБйЗЗиі≠дЊЫеЇФгАБжЦљеЈ•еЃЙи£ЕгАБиѓХиљ¶й™МжФґгАБеЈ•з®ЛињЫеЇ¶гАБжКАжЬѓиі®йЗПгАБеЃЙеЕ®зОѓдњЭгАБиµДйЗСињРдљЬгАБйГ®йЧ®еНПи∞Гз≠ЙжПРдЊЫдЇЖиѓ¶е∞љзЪДиІДиМГи¶Бж±ВпЉМеИґиНѓдЉБдЄЪеЇФиЗ™иІЙдї•еЫљж†ЗGB/T 25915дЄЇеЗЖзї≥пЉМеєґжНЃж≠§еИґиЃҐеМїиНѓеЈ•з®Лй°єзЫЃзЪДGEPпЉМз°ЃдњЭеЈ•з®Лй°єзЫЃзђ¶еРИ GMP и¶Бж±ВгАВ
еЫљж†ЗGB/T 25915з≥їеИЧж†ЗеЗЖеПНжШ†дЇЖељУдїКеЫљйЩЕвАЬжіБеЗАеЃ§еПКзЫЄеЕ≥еПЧжОІзОѓеҐГ вАЭзЪДжЦ∞ж¶ВењµгАБжЦ∞жАЭиЈѓеТМжЦ∞жЦєж≥ХпЉМжШѓжМЗеѓЉжИСеЫљеРДи°МеРДдЄЪжіБеЗАжКАжЬѓзЪДиІДиМГжАІжЦЗдїґпЉМењЕе∞ЖжИРдЄЇжИСеЫљжіБеЗАжКАжЬѓиУђеЛГеПСе±ХзЪДжЦ∞иµЈзВєгАВ
жЬђжЦЗж†Зз≠ЊпЉЪжіБеЗАеЃ§
еЬ∞еЭАпЉЪе±±дЄЬзЬБжµОеНЧеЄВ姩氕еМЇжЦ∞еЊРе±ЕеІФдЉЪйїДж≤≥еїЇйВ¶е§Іж°•и•њдЊІ1-6еПЈ зФµиѓЭпЉЪ0531-68824415 дЉ†зЬЯпЉЪ0531-68824415
зЙИжЭГжЙАжЬЙ:жµОеНЧй°Їе•ЗеЗАеМЦеЈ•з®ЛжЬЙйЩРеЕђеПЄ жКАжЬѓжФѓжМБпЉЪеЇЈзЊОзІСжКА е§Зж°ИеПЈ:й≤БICPе§З19040779еПЈ xmlеЬ∞еЫЊ htmlеЬ∞еЫЊ txtеЬ∞еЫЊ зЩїељХ
